A 37-year-old male applied for life insurance. He has a history of metastatic paraganglioma initially diagnosed in September 2009 after symptoms of left chest and left groin pain and fatigue, followed by a finding on CT scan, ultrasound and MRI of an 18x16x8.5 cm tumor in the left perirenal area.
A genetic test revealed a mutation in SDHB (succinate dehydrogenase unit B) gene. In 2010, he was found to have metastasis to the bones, ribs, sternum, left upper abdomen and lungs. He received treatment with 135 mCI of 131 iodine MIBG. Seven years later he had a CT scan of the chest, abdomen and pelvis that did not show evidence of progressive disease. But a slight elevation of chromogranin A prompted an MRI of the cervical, thoracic and lumbosacral spine which showed two lesions at the level of T3 and T4 not seen previously. A PET scan was ordered, but the results are not provided.
What is a paraganglioma tumor and what are the mortality implications?
Definition
Paragangliomas (PGLs) are rare vascular, neuroendocrine tumors of paraganglia cell clusters originating from the neural crest that have co-migrated with the autonomic nervous system. PGLs are associated with either the sympathetic tissue in adrenal (pheochromocytomas (PCCs)) and extra-adrenal locations (sympathetic PGLs (sPGLs)) or the parasympathetic tissue of the head and neck paragangliomas (parasPGLs, formerly called glomus tumors).
PCCs and sPGLs are catecholamine-secreting tumors that arise from chromaffin cells, whereas parasPGLs are mostly chromaffin-negative tumors and usually non-functioning.
Etiology
Most PGLs occur as sporadic tumors, however at least one-third of PGLs/PCCs are hereditary. Four hereditary syndromes can be associated with development of PGLs:
Von Hippel-Lindau Syndrome: patients with VHL gene (found on chromosome 3) can develop pheochromocytomas (often bilateral), paragangliomas, retinal angiomas, central nervous system hemangioblastomas, renal cell carcinoma, renal and pancreatic cysts, pancreatic endocrine tumors and epididymal cystadenoma.
Multiple Endocrine Neoplasia Syndrome type 2A and 2B: are due to mutations in the RET gene and usually associated with bilateral pheochromocytomas.
Neurofibromatosis type 1, also known as Von Recklinghausen’s disease: the result of mutation in chromosome 17; pheochromocytomas can occur in up to 5% of cases, both inside and outside the adrenal gland.
Familial Paraganglioma Syndrome: is caused by mutations in the succinate dehydrogenase (SDH) gene and can be associated, albeit rarely, with pheochromocytomas.
Epidemiology
The prevalence of PGLs is unknown, but has been estimated to be between 1:6500 and 1:2500 in the USA. The annual incidence has been reported to be two to 10 cases per million. PGLs may occur at all ages, with the highest incidence between 40 and 50 years, and with an approximately equal sex distribution. Most PGLs/PCCs are benign, but 10% to 15% are malignant, defined by the presence of metastatic spread in sites where chromaffin tissue is normally absent, such as lymph nodes, liver, bone and lungs.
Malignant pheochromocytoma and sympathetic paraganglioma affect only about 100–200 people per year in the United States.
Figure 1 — CT scan showing tumor
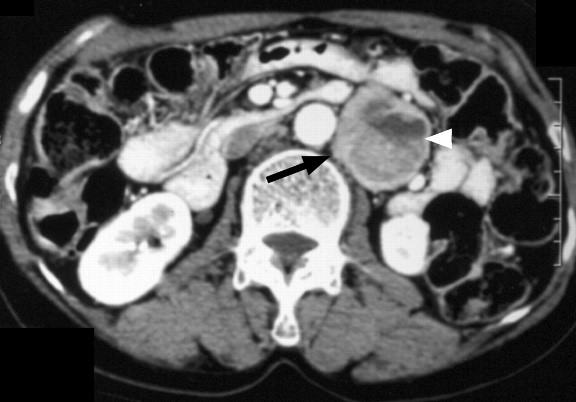
Enhanced CT scan show 4.5-cm retroperitoneal PGL (arrow) with heterogeneous enhancement in left paraaortic area. Note large cystic component with fluid level (arrowhead) within tumor mass. High attenuation of dependent fluid in cystic portion is suggestive of intratumoral hemorrhage. Source: American Journal of Roentgenology. 2006;187:492+
Diagnosis
The diagnosis of paragangliomas is based on physical examination, biochemical testing, and imaging studies. Pheochromocytomas in the adrenal gland may produce both adrenaline and noradrenaline. However, only paragangliomas that occur in the organ of Zuckerkandl, which is found along the lower part of the aorta, can make adrenaline. The remainder of paragangliomas are either non-functioning or produce noradrenaline only.
Figure 2 — Paragangliomas (PGL) and Incidence of malignancy by PGL type
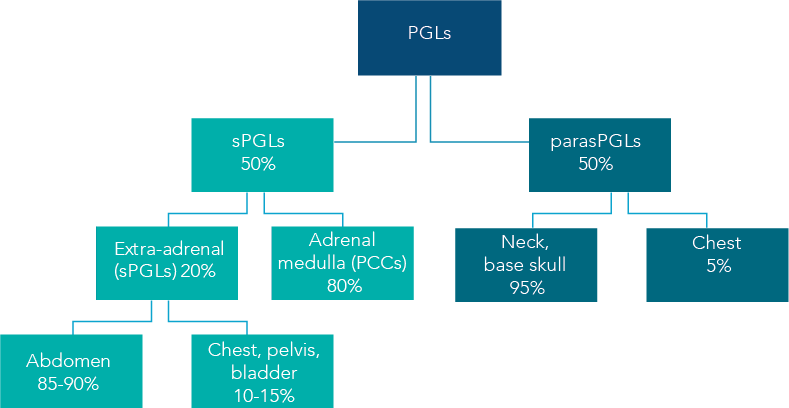
Paragangliomas (PGLs) are rare vascular, neuroendocrine tumors of paraganglia cell clusters originating from the neural crest that have co-migrated with the autonomic nervous system. The chart at right shows the incidence of malignancy of various types of PGLs.
Clinical presentation
The majority of sPGLs and PCCs produces catecholamines in advanced cases causing symptoms of catecholamine excess. Highly variable symptomatology in patients with PGLs may reflect variations in the nature and types of catecholamines secreted, as well as co-secretion of neuropeptides. Hypertension, continuous or paroxysmal, is the most common feature of advanced PCCs and sPGLs. Typical symptoms are paroxysms of severe headache, palpitations and diaphoresis, ‘the classic triad’. Other symptoms may include anxiety, nausea, vomiting and weakness. In addition, hyperglycemia, resulting from metabolic actions of catecholamines, may be the presenting symptom.
ParasPGLs are usually benign and only a minority (4%) produces catecholamines. They can remain clinically silent for years. But their usual location in the head and neck in the proximity of nerves and vascular structures often results in considerable morbidity due to compression or infiltration of the adjacent structures. They may cause symptoms such as hearing loss, tinnitus, dysphagia and cranial nerve palsy. Carotid body tumors are the most common parasympathetic PGL, usually presenting as a painless neck mass.
Biochemical tests
Studies have found the rate of elevated catecholamine metabolites to be 88% in abdominal/pelvic PGL. Compared to PCC the levels of urinary metanephrines, while elevated, were much lower. The level of normetanephrines in PGL were much higher than in PCC. Levels of catecholamines (adrenaline, noradrenaline, dopamine) and metanephrines may be measured in either the plasma or in a 24hour urine collection. Levels that are 2 or more times the upper limit of normal levels confirm the diagnosis of paraganglioma.
Figure 3— Retrospective Study Results
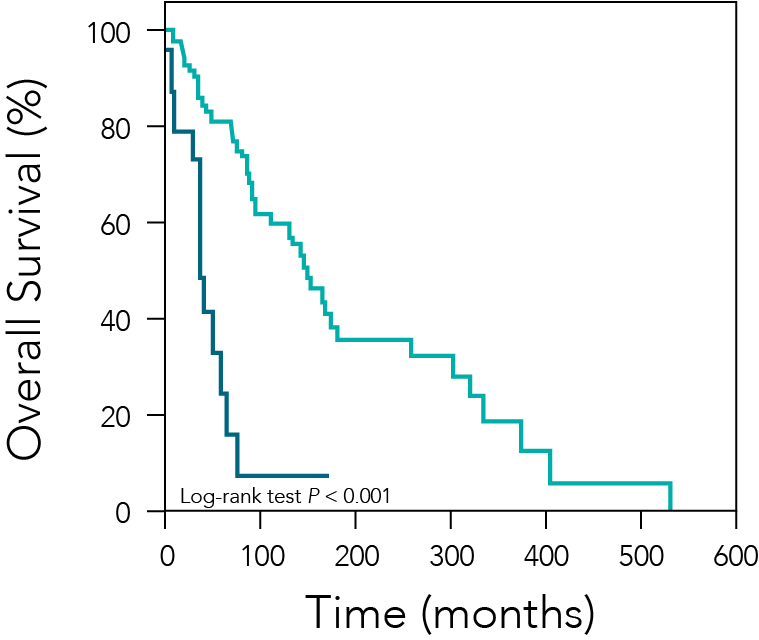
In a retrospective study, patients with metastatic pheochromocytoma or paraganglioma who underwent palliative resection of the primary tumor (green line, n = 28) had longer median overall survival than those who did not (blue line, n = 24). Roman-Gonzalez A, et al. Ann Surg. 2017
Imaging studies
When the diagnosis is confirmed with plasma or urine studies, the patient should have a CT scan or MRI to identify the location of the PGL. Nuclear medicine tests like a MIBG (metaiodobenzylguanidine) scan or PET (positron emission tomography) scan can be helpful in locating tumors. The MIBG scan is specific for adrenaline-producing tumors while the PET scan identifies any very metabolically active tumors.
There is no definite histological finding that can be used for the diagnosis of malignant PGL. Therefore, malignancy is defined by the presence of metastases: tumor spread to sites where chromaffin tissue is normally absent.
Natural history
These tumors are derived either from sympathetic tissue in adrenal or extra-adrenal abdominal locations (sPGLs) or from parasympathetic tissue in the thorax or head and neck (parasPGLs); the two types occur with similar frequency.
sPGLs may develop anywhere there are sympathetic nerve cells, and this usually means along any of the major arteries in the body. They frequently produce considerable amounts of catecholamines, and in approximately 80% of patients, they are found in the adrenal medulla.
The remaining 20% of these tumors are located outside of the adrenal glands, in the prevertebral and paravertebral sympathetic ganglia of the chest, abdomen, pelvis and bladder. Far and away, the most common site is within the abdomen where approximately 85-90% are located. Most abdominal sPGLs arise from a collection of chromaffin tissue around the origin of the inferior mesenteric artery (the organ of Zuckerkandl) or aortic bifurcation.
In contrast, the most common location for a parasPGL is the carotid body in the neck. Carotid body paragangliomas arise at the bifurcation of the internal and external carotid arteries and have classic radiographic features.
While most PPGLs are benign, about 10% of PCCs, 20-25% of extra-adrenal abdominal and mediastinal secretory PGLs are malignant. In the skull base and neck, malignancy is least common for jugulo-tympanic tumors (2-4%), slightly higher for carotid body tumors (4-6%), and highest for vagal tumors (10-19%).
Compared to adrenal PCCs, PGLs have a higher risk of being malignant or cancerous, as high as 40-50% in some studies. Larger tumor size (over 5 cm), invasion of the tumor into adjacent structures, and metastases to other organs are characteristics of malignant paragangliomas. PGLs typically metastasize to lungs, liver, bones, and lymph nodes.
Paraganglioma syndrome 4 (PGL4) is associated with mutations in SDHB at gene locus 1 and is the second most common type of familial paraganglioma. SDHB mutations are associated with a higher malignancy rate (21% to 79%) than other types of SDHx-associated familial paraganglioma syndromes, and with renal cell carcinoma.
Treatment
The definitive treatment of PGLs is surgical excision of the tumor. Laparoscopic surgery is commonly the technique of first choice for resection adrenal and extra-adrenal PGLs when oncologic principles can be followed. Surgery can also be used as a curative treatment for recurrent or limited metastatic tumors.
Traditional chemo-therapy with cyclophosphamide, vincristine, and dacarbazine (CVD) has been used most extensively with progressive and widely metastatic PGLs. Molecular targeted therapies such as sunitinib (tyrosine kinase inhibitor) and everolimus (mTOR inhibitor) have been tried with mixed results.
Prognosis
The long-term prognosis of patients after operation for non-malignant PGLs is excellent, although nearly 50% may remain hypertensive after surgery. On long-term follow-up, about 17% of tumors recur, with about half of these showing signs of malignancy. Although follow-up is especially important for patients identified with mutations of disease-causing genes, there is currently no method based on pathological examination of a resected tumor to rule out potential for malignancy or recurrence. The prognosis of metastatic PCC/PGLs is variable. Long-term survival is possible even in the presence of distant metastatic disease, but five-year survival rates are ≤ 60%.
Prognosis is impacted by tumor burden, location of metastases, and rate of progression; patients with brain, liver and lung metastases tend to have a worse prognosis than do those with isolated bone lesions.
Returning to the case
Unfortunately, and despite the resection of the primary PGL tumor with the nephrectomy, this case has unfavorable features such as the onset of a malignant PGL at a relatively-young age, the very large dimensions of the tumor, and the presence of metastases. Moreover, the two lesions recently found on the MRI of his spine are quite possibly a recurrence. Finally, the mortality rate associated with metastases would raise very significant long-term mortality concerns.
References
Roman-Gonzalez A, Jimenez C. Malignant pheochromocytoma-paraganglioma: pathogenesis, TNM staging, and current clinical trials. Curr Opin Endocrinol Diabetes Obes. 2017;24: 174–183.
Bronson S. Malignant Pheochromocytoma and Sympathetic Paraganglioma Research OncoLog, August 2017, Volume 62, Issue 8
Corssmit E.P. and Romijn J.A. Clinical management of paragangliomas European Journal of Endocrinology (2014) 171, R231–R243
Vasiliu et al Malignant Paraganglioma; a Story of a Long Time Survival J Surgery 2015 Volume 10, Issue 2-15.
Prades C.A.et al Metastatic Malignant Paraganglioma: A Case Report and Review of Literature World J Oncol. 2017;8 (3):92-95
Fliedner S.M.J. et al Metastatic Paraganglioma Semin Oncol. 2010 December; 37(6): 627–637.
Tischler A.S. and deKrijger R.R. Pathology of pheochromocytoma and paraganglioma Endocrine-Related Cancer (2015) 22, T123–T133
Pacak K. and Tella S.H Pheochromocytoma and Paraganglioma (2018) Endotext - NCBI Bookshelf.
Canu L et al Sunitinib in the therapy of malignant paragangliomas: report on the efficacy in a SDHB mutation carrier and review of the literature Arch Endocrinol Metab. 2017;61/1.